 2015, Vol. 46
2015, Vol. 46中国海洋湖沼学会主办。
文章信息
- 刘晓辉, 王健鑫, 王帅兵, 樊英萍, 俞凯成, 蒋然, 刘明华. 2015.
- LIU Xiao-Hui, WANG Jian-Xin, WANG Shuai-Bing, FAN Ying-Ping, YU Kai-Cheng, JIANG Ran, LIU Ming-Hua. 2015.
- 长江口及邻近海域表层海水细菌多样性及群落结构
- BACTERIAL DIVERSITY AND COMMUNITY STRUCTURE IN SURFACE SEAWATER OF CHANGJIANG RIVER ESTUARY AND ADJACENT AREAS
- 海洋与湖沼, 46(6): 1531-1541
- Oceanologia et Limnologia Sinica, 46(6): 1531-1541.
- http://dx.doi.org/10.11693/hyhz20150900243
-
文章历史
- 收稿日期:2015-09-17
- 改回日期:2015-09-28




海洋约占地球表面积的71%,浩瀚的海洋中有着丰富的生物资源,而海洋微生物以其数量大、种类多、分布广及适应能力强等特点在其中占据举足轻重的地位。特定环境下的微生物群落结构不仅可以反映微生物所处的生境特点,同时也能说明微生物在生态系统中的功能及其在生物地球化学循环中所起的作用(Lu et al,2008),因此通过分析自然环境中的细菌群落结构来研究生态学、环境学及全球变化,有着重要的现实意义。
长江是我国第一大河,平均年入海径流量约9.28×1011 m3,占输入东海总径流量的84.4%,在陆源物质向东海的输送中起着重要作用(张东声,2011)。大量营养盐、污染物等输入长江口及东海,对河口和沿岸海域的水文、沉积过程、地貌变化、生物生态等产生重要影响,另外长江口及邻近海域还受到台湾暖流、浙江近岸流和黄海沿岸流等水系的共同作用(Ning et al,2011),强烈的季风气候通过海气相互作用也影响东海的环流和温盐结构,这种多水系的汇合、复杂的水文状况和日益频繁的人类活动干扰,使该海域不仅是我国海岸带陆-海相互作用研究的关键水域(宁修仁等,2004),也是典型近岸生态系统研究的重要区域。
近年来,关于长江口及邻近海域微生物多样性的研究日益增多,Jiao等(2007)应用16S rRNA 基因文库的方法研究了秋季长江口和东海陆架区的微生物多样性,刘敏等(2008)采用PCR-DGGE 技术对长江口外低氧区的细菌群落组成进行了分析,曾永辉(2008)和潘洛安(2005)也分别研究了长江口及邻近海域浮游细菌多样性和分布特征,但总体由于采样站点、季节、方法的差异,对该海域细菌组成及群落结构仍然缺乏比较全面的认识。
本文采用DAPI 染色计数法对长江口及邻近海域10 个站点表层海水中的细菌生物量进行统计,并结合16S rRNA 基因克隆文库法,构建系统发育树,研究了长江口及邻近海域细菌群落结构及其多样性,为了解海洋微生物多样性提供了分子生物学方面的基础数据,也为研究海洋细菌与生态系统之间的关系提供一定的参考。
1 材料与方法1.1 样品采集2014 年7 月,搭乘“浙海科1 号”科学考察船,利用SBE 32 型采水器(海鸟公司),在长江口及邻近海域共采集10 个站点表层海水样品,其采样站点分布如图 1 所示。

|
| 图 1 长江口及邻近海域采样站点分布图 Fig. 1 The sampling sites of the Changjiang River estuary and adjacent areas |
样品采集 采集表层(水面下2m)海水30mL,经100μm的筛绢过滤后,分装至10mL 的无菌冻存管中,立即加浓度为1%的戊二醛固定液进行固定,黑暗固定15 分钟后放入低温冰箱的冷冻格(-20°C)中保存。
1.1.2 环境总DNA提取样品采集 采集表层海水1000mL,装于灭菌的聚乙烯塑料瓶中。样品先用100μm 的筛绢过滤,再经孔径为3μm和0.22μm 的混合纤维素滤膜(Millipore 公司)过滤,将过滤后的0.22μm 滤膜放入灭菌的5mL 冻存管中,置-20°C 冰箱中保存,航次结束后放置于实验室-80°C 超低温冰箱中长期保存。
1.2 DAPI 染色计数取1—2mL 待测水样,加入终浓度为10μg/mL 的DAPI 染色剂,室温下暗房染色30 min,加入含0.2μm黑色聚碳酸酯膜的过滤器,滤干后取出滤膜,放在干净的载玻片上展平,在滤膜中间滴一滴香柏油,置于荧光显微镜(100 倍油镜)下观察并随机选择10 个视野进行计数,只计算清晰可见且为蓝光的菌体。根据公式BN=A×S1/(S2×V)计算每毫升细菌数,其中BN 为每毫升细菌的数量,A 为视野中细菌的平均数,S1 为滤膜的面积,S2 为显微镜的视野面积,V 为过滤水样的体积(赵海萍等,2007)。
1.3 环境总DNA 提取和16S rDNA 扩增利用FastPrep 快速核酸提取仪(MP Biomedicals公司)和FastDNA spin kit for soil 试剂盒进行海水样品总DNA 提取,核酸蛋白检测仪(Bio-Rad 公司)测定DNA 浓度和纯度,纯化后用于PCR 扩增。扩增采用引物341f(5'-CCTACGGGAGGCAGCAG-3')和907r(5'-CCGTCAATTCCTTTGAGTTT-3')。50 μL 反应体系为: 10×buffer 5 μL,dNTPs 0.5 μL,Primer 341f 1μL,Primer 907r 1 μL,H2O41 μL,Taq 酶(TaKaRa)0.5 μL,模板DNA 1 μL。PCR 扩增条件为: 95°C 预变性5 min,94°C 变性1 min,56°C 退火30 s,72°C 延伸2 min,循环35 次,最后72°C 延伸10 min。
1.4 细菌克隆文库构建及序列分析PCR 回收产物与pMD-18T vector(TaKaRa 公司)在16°C 连接过夜,将连接产物转化到大肠杆菌DH5α感受态细胞(TaKaRa 公司),经蓝白斑筛选,挑选阳性克隆,重新扩增插入片断,将含有合适大小插入片段的克隆送苏州金唯智生物科技有限公司进行测序。获得序列后,首先利用Usearch 软件(http: //drive5.com/usearch/)去除嵌合体,再应用BLASTn 程序搜索相似性序列,进行系统发育分析。采用ClustalX(Version1.8)对序列进行比对分析,通过MEGA 5 软件构建系统发育树(Kumar et al,2004),采用Neighbor-Joining建树方法,建树结果进行Bootstrap1000 次系统检验,利用PHYLIP 软件包中DNASIS 程序计算距离矩阵,利用DOTUR 软件将相似性>97%的序列归为一个OTU,计算多样性指数并绘制稀释曲线(Schloss et al,2005)。
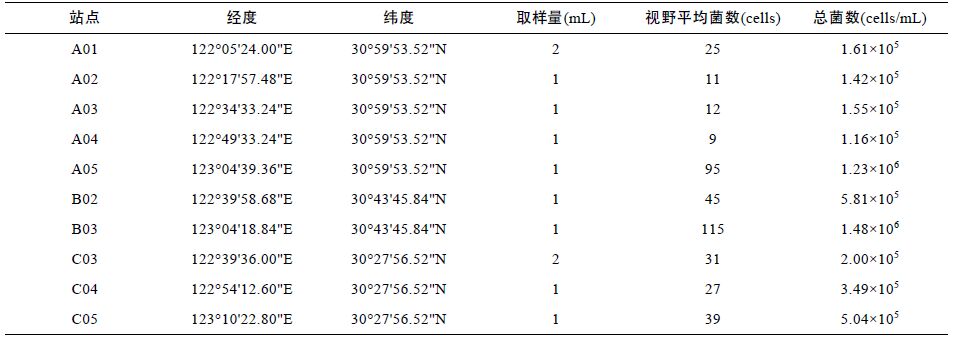
2 结果与分析2.1 DAPI 染色计数DAPI 染色计数结果如表 1 所示,其中B03 站点总菌数(1.48×106 cells/mL)最高,其次为A05 站点(1.23×106 cells/mL),而A04 站点的总菌数(1.16×105 cells/mL)最低,10 个站点的平均值为4.92×105 cells/mL,细菌丰度总体较高,但各站点之间细菌丰度也存在明显差异,细菌丰度的高值区主要集中于舟山外海一带,而低值区主要集中于长江口区域,整体变化趋势为从长江口至外海区域细菌丰度逐渐增加。
 |
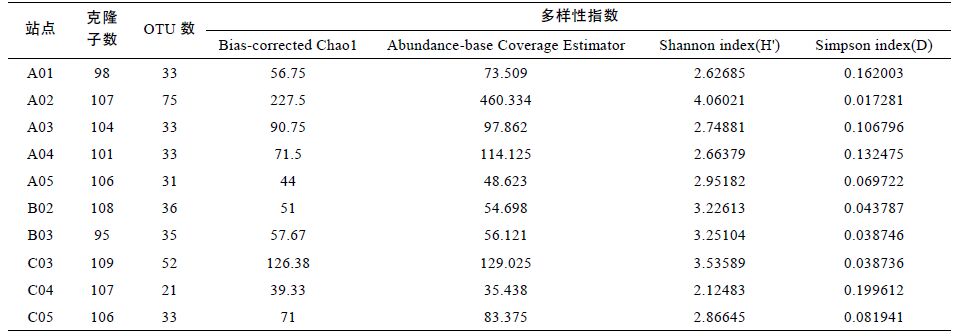
每个站点选取110 个左右阳性克隆进行测序,片段长度为550—700bp; 利用DOTUR 软件,将相似性>97%的克隆子归为一个OTU,各站点的克隆文库基本情况及多样性指数如表 2 所示,细菌群落的稀释曲线如图 2 所示。
 |

|
| 图 2 十个文库的稀释性曲线 Fig. 2 The rarefaction curve of the 10 clone libraries |
Chao1和ACE 指数用于计算群落的物种丰富度,在不同算法的计算下它们有着相同的变化趋势。由表 2 可知,丰富度指数Chao 1 与ACE指数同时显示A02站点拥有最高丰富度,C03 站点次之,丰富度最低的为C04 站点。Shannon和Simposon 指数用于计算群落的多样性,由于计算方法的差异它们在数值上的变化趋势是相反的,但两者均显示A02 站点细菌多样性最高,C03 站点次之,C04 站点最低。结合各站点的群落丰富度和多样性指数,可以看出A02 站点的细菌群落多样性最高。由稀释性曲线可知,A02和C03站点的稀释曲线仍然有上升的趋势,表明该站点中细菌群落多样性很高,克隆子数量还未完全覆盖其多样性,这与其丰富度和多样性指数结果是一致的,其余站点的稀释性曲线均趋于平稳,基本上能反映真实环境中细菌的群落结构情况。
2.2.2 细菌克隆文库物种类群分析通过BLASTn 搜索相似序列,可将10 个站点共1041 个克隆子分为11 个细菌门类,分别为变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)、放线菌门(Actinobacteria)、蓝细菌门(Cyanobacteria)、厚壁菌门(Firmicutes)、绿弯菌门(Chloroflexi)、酸杆菌门(Acidobacteria)、浮霉菌门(Planctomycetes)、纤维杆菌门(Fibrobacteres)、疣微菌门(Verrucomicrobia)和柔膜菌门(Tenericutes),另有2.02%的细菌分类地位不确定。变形菌门又分为α-变形菌纲(Alphaproteobacteria)、β-变形菌纲(Betaproteobacteria)、γ- 变形菌纲(Gammaproteobacteria)、δ-变形菌纲(Deltaproteobacteria)和ε-变形菌纲(Epsilonproteobacteria)五个纲。各细菌类群在每个站点克隆文库中的比例如图 3 所示。

|
| 图 3 细菌类群在不同采样点克隆文库中的比例 Fig. 3 The ratios of bacterial groups in clone library of different sites |
综合10 个站点文库的结果可以发现,变形菌门为研究区域中最大的优势菌群(占60.61%),其中α-变形菌占50.62%,β-变形菌占4.32%,γ-变形菌占4.13%,δ-变形菌占1.44%,ε-变形菌占0.10%; 仅次于变形菌门的是拟杆菌门(占15.18%); 第三大优势菌群为放线菌门(占14.79%)。
对各站点文库进行比较分析,可见除B03 站点外,变形菌门是其余9 个站点文库的主要优势类群,其中C04 站点文库比例最高(占90.66%),变形菌门中又以α-变形菌为主要类群,其余类群(特别是δ-变形菌和ε-变形菌)所占比例相对较少,ε-变形菌仅在A02站点有发现且含量很低(仅占0.93%)。作为第二大优势类群的拟杆菌,仅在B03 站点克隆文库中的含量超过变形菌门(占43.16%),成为该站点文库的主要类群,其它站点均低于变形菌门。放线菌门在10 个站点克隆文库中均有分布,其中A02 站点放线菌含量最高(占35.51%),与该站点的变形菌门含量相当。
除了A01 站点未见相应克隆,蓝细菌门在各文库的分布比较均匀,但含量不高,最高值出现在C05 站点(占12.26%)。浮霉菌门只在A02和C03 站点文库中有分布,并且含量极低(分别为0.93%和0.92%)。靠近长江口的A01和A02 站点文库含有其它站点没有的厚壁菌门,其含量分别占各自文库的8.16%和3.74%。绿弯菌门主要分布于A 断面(A01、A02、A03和A04 站点),其中A02 文库中绿弯菌门含量最高(占3.74%)。柔膜菌门仅出现在C05 站点文库中,且仅占0.94%。A04 站点文库含有其它站点文库没有的纤维杆菌门(占1.98%)。酸杆菌门和疣微菌门只在A02 站点的克隆文库中出现,含量分别为2.80%和1.87%。同时分类地位不确定的类群在各站点文库中都有极少量的分布。
2.3 细菌系统发育分析 2.3.1 变形菌门系统发育分析使用MEGA5.0 软件,对各站点典型变形菌序列与BLASTn 比对相似序列构建系统发育树(图 4),克隆子相似菌株分离源的分析结果如表 3 所示。

|
| 图 4 根据细菌16S rDNA 基因序列构建的变形菌系统发育树 Fig. 4 The phylogenetic tree of Proteobacteria according to 16S rDNA gene sequences |
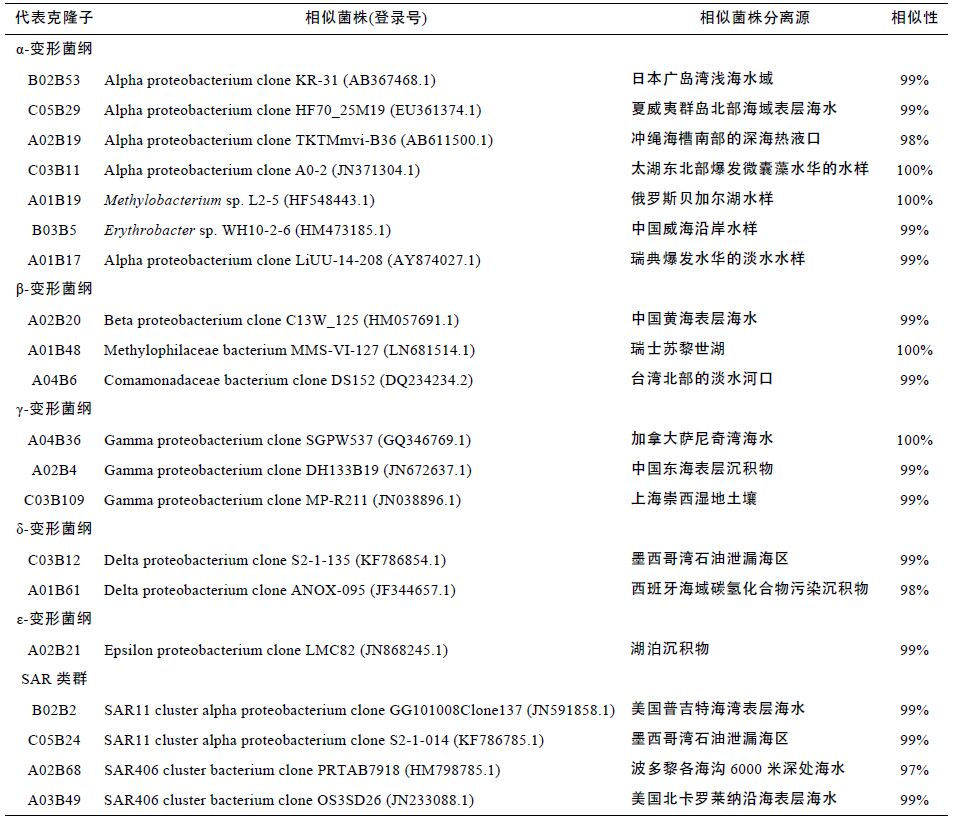 |
由系统发育树和相似序列生境分析结果可知,α-变形菌纲克隆子的相似序列主要来自表层海水、富营养化淡水水域,个别源自深海热液口; β-变形菌纲克隆子的相似序列主要来自淡水湖泊和海水环境; γ-变形菌纲克隆子的相似序列主要来自表层水体、海洋沉积物和土壤环境; δ-变形菌纲克隆子相似序列的分离源主要是墨西哥湾石油泄漏海区和西班牙海域碳氢化合物污染的沉积物,推测该菌可能与石油烃污染环境相关; ε-变形菌则在水体环境中分布较少,唯一的基因型A02B21 与湖泊沉积物中的ε-变形菌同源性达99%。SAR 类群是海洋中比较典型的细菌类群,也是影响海洋碳循环的重要因素,由图表可知,SAR11的相似菌株来自美国普吉特海湾表层海水和墨西哥湾石油污染海域; SAR406 的分离源则分别来自沿海表层和深海的海水环境,说明该类群细菌对于压力、光照等环境具有很强的适应性。
2.3.2 其它细菌类群系统发育分析除变形菌门外,各站点其它典型细菌序列与BLASTn 比对相似序列所构建的系统发育树如图 5 所示,克隆子相似菌株分离源的分析结果见表 4。

|
| 图 5 根据细菌16S rDNA 基因序列构建的其它分支细菌系统发育树 Fig. 5 The phylogenetic tree of other bacteria according to 16S rDNA gene sequences |
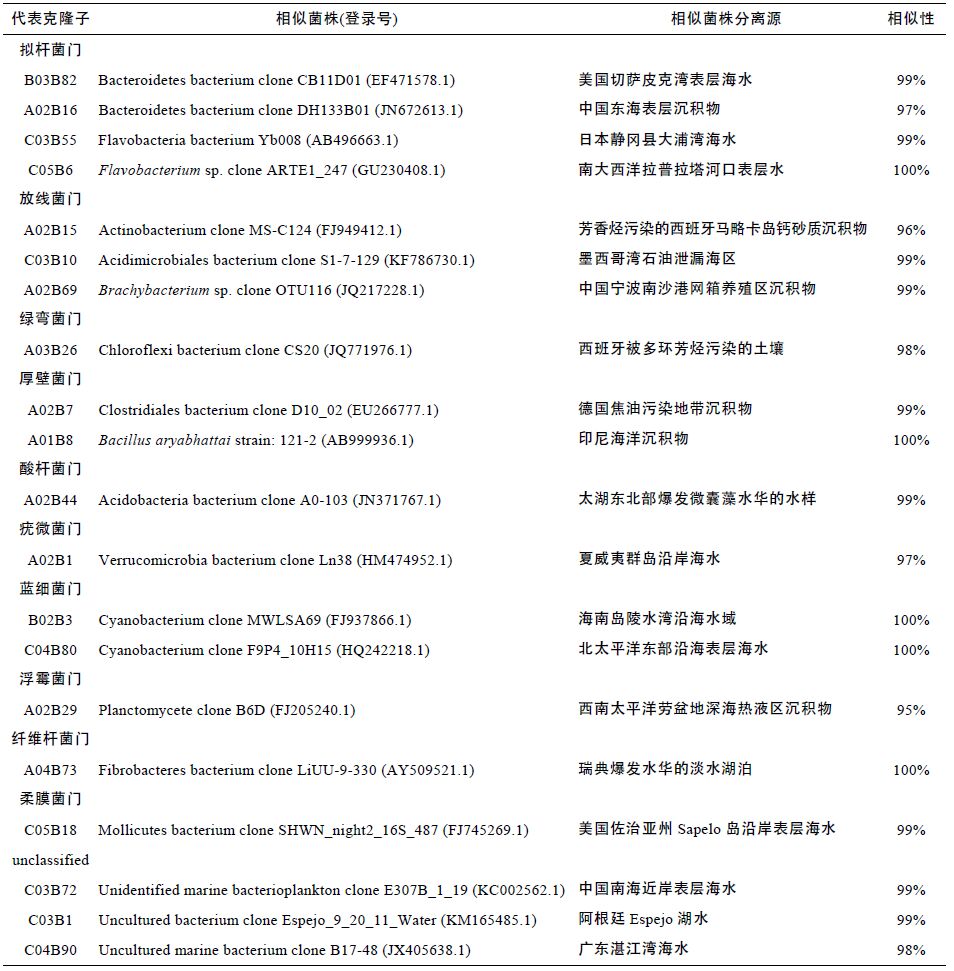 |
由系统发育树的结果可知,变形菌门以外的其它代表性细菌克隆,其相似序列大多来自海水环境,少数来源于沉积物和淡水环境。其中拟杆菌门(黄杆菌纲)克隆子的相似序列主要来自于河口和海湾等生境; 放线菌门、绿弯菌门、厚壁菌门等细菌克隆子的相似菌株则大多来源于受芳香烃、石油、多环芳烃等污染的海域、土壤或沉积物; 酸杆菌门克隆子的相似序列来自太湖东北部爆发微囊藻水华的水样; 疣微菌门的相似序列则来自夏威夷群岛沿岸海水; 蓝细菌门克隆子相似序列的分布比较广泛,主要来自各海域的表层海水; 浮霉菌门克隆子相似序列的生境来自西南太平洋劳盆地深海热液区沉积物;另外分布较少的纤维杆菌门和柔膜菌门,其克隆子相似序列分别来自瑞典爆发水华的淡水湖泊和美国佐治亚州Sapelo 岛沿岸表层海水。在分类地位不明确的细菌中,基因型B02B83(1 个克隆子)、C03B1(4个克隆子)、和C04B90(1 个克隆子)在发育树中处于同一个单独的分支,其相似序列主要来自我国南海近岸表层海水、阿根廷Espejo 湖水以及广东湛江湾海水。
3 讨论3.1 长江口及邻近海域表层海水细菌丰度从长江口到舟山外海的区域是受人类活动影响最大的区域,每年有着大量的陆源有机物质输入,而该区域又是台湾暖流和长江冲淡水交汇的区域(唐晓晖等,2004),复杂的水文地理条件使得该区域的细菌丰度同时受众多环境因素的影响。赵三军等(2003)研究发现秋季长江口外海域表层海水的细菌丰度为3.97×105—1.162×106 cells/mL 之间,平均值为7.19×105 cells/mL; 刘晶晶等(2011)调查了夏季长江口及邻近海域浮游细菌的数量,结果为6.92×105—5.54×106cells/mL 之间; 王新等(2010)对春季东海赤潮高发区的浮游细菌丰度调查结果为5.85×104—9.26×105cells/mL 之间; 上述研究结果与本次调查结果(细菌丰度在1.16×105—1.48×106cells/mL 之间,平均值为4.92×105cells/mL)都有一定差异,原因主要可能是采样季节不同以及环境特征的改变。
就细菌丰度的空间变化而言,本研究发现细菌丰度分别在A05、B03、C05等靠近外海的站点形成高值区,在相对离岸较近的A01—A04和C03—C04站点海域形成低值区,细菌丰度的总体空间变化趋势是外海较近岸高,其主要原因可能是外海区域的几个站点位于营养盐丰富、光照充足的高生产力海域,浮游植物的繁盛促进了细菌的大量生长; 相反,在长江口近岸海域,由于水团之间混合所产生的高浊度和低透光率影响了浮游植物生长,从而形成海洋细菌低密度区。季倩(2008)研究认为长江口靠近最大混浊带异养细菌丰度较低,由长江口向外海其丰度显著增加; 宁修仁等(2004)也认为在长江口及邻近区域的冲淡水区中部以及冲淡水和海水交汇处是低细菌丰度和活性区域,上述报道与本次的研究结果基本一致。本次研究中B02 站点虽然处在A03和C03站点相近经度区域,但细菌生物量处于较高水平,原因可能是B02 站点位于拥有大量天然海藻场和人工贻贝养殖场的枸杞岛海域,养殖区域的存在对细菌丰度的影响较大,有研究表明浮游植物和海藻光合作用产物以及部分海藻降解产生的腐殖质促进了浮游细菌的新陈代谢(Vörös et al,2003),同时贝类养殖的残饵、粪便所分解的有机质含量较高,在该海域独特的水文地理条件下可能导致水体中细菌丰度偏高。
3.2 长江口及邻近海域细菌多样性及群落结构近年来国内外有许多针对长江口海域表层海水细菌多样性及群落结构的相关研究,如李和阳等(2012)研究了长江口典型站位浮游细菌类群的组成特征,结果表明变形菌门(α-,β-和γ-变形菌纲)为该海域优势菌群; Feng等(2009)对长江口及邻近海域海水和沉积物样品进行较全面的研究,结果显示变形菌门是该海洋环境中含量最丰富的种类(占克隆数的72.9%),其中α-变形菌纲和γ-变形菌纲为该海域海水中的最大优势菌群; Sekiguchi等(2002)分析了长江口特定站位细菌的遗传多样性,发现优势菌为α-变形菌和γ-变形菌。从以上研究结果可以发现变形菌门是长江口及邻近海域表层海水中的优势菌群,作者在本实验中检测到变形菌门含量达到总克隆数的60.61%,是本次研究海域表层海水中的最大优势类群,这与以往研究结果基本一致。变形菌门作为本实验的优势类群,在长江口及邻近海域生态系统中发挥着重要作用,尤其是含量丰富、分布广泛的α-变形菌纲(占总克隆数的50.62%)。目前对α-变形菌的研究发现,其在低价硫化物的转化过程中起间接作用,常以H2S 作为代谢中的电子供体(李涛等,2008); 多数α-变形菌还具有广泛的耐盐性,并且利用DOC等碳源的能力很强(Sjöstedt et al,2012)。本实验中有5 个站点的α-变形菌含量高达50%以上,说明相应环境特征非常适合该细菌类群的生长,同时也验证了从长江口到外海区域盐度和营养盐急剧变化条件下α-变形菌仍为本次研究海域的优势菌群。β-变形菌是世界范围内淡水湖泊和河口区域的优势菌群(Mueller-Spitz et al,2009; Cottrell et al,2005),但本次研究中β-变形菌含量仅占总文库的4.32%,这可能与长江口冲淡水的影响有关。γ-变形菌是海水和沉积物环境中的常见细菌类群,与光能营养、趋碱性、氨氧化和硫降解等过程相关(Bowman et al,2005;Ravenschlag et al,1999),目前较多报道显示γ-变形菌在沉积物环境中属于优势类群(Bowman et al,2003;Inagaki et al,2003),但在海水环境中不是优势菌群,我们的研究结果也验证了这一观点。δ-变形菌常在硫循环以及铁等金属的代谢中扮演重要角色(Xu et al,2008),本次研究发现δ-变形菌克隆与原油泄漏海域的脱硫杆菌相似度高,脱硫杆菌是一种能降解有机物(尤其是石油烃类)的硫酸盐还原细菌(Musat et al,2009),这预示着δ-变形菌含量丰富的站点可能受到石油烃的污染。ε-变形菌虽然能广泛利用环境中的氢、硫化物、硝酸盐、氧气等多种物质作为电子受体或供体(Takai et al,2003; Nakagawa et al,2005),但在海水中的分布极少,本次调查也仅在A02 站点有发现。
拟杆菌门、放线菌门和蓝细菌门是本次研究分布比较广并且比例也较高的类群。本次实验发现的拟杆菌主要包括黄杆菌纲和鞘脂杆菌纲两大类,其中黄杆菌纲与藻类的赤潮和降解过程有关(Abell et al,2005),而鞘脂杆菌纲重要类群为噬胞菌属(Cytophaga)可以降解纤维素(Li et al,2011),上述拟杆菌门含量较高与该海域初级生产力水平有关。放线菌是一类在海洋中广泛存在且丰度较高的类群,有研究表明放线菌在有机污染物的降解过程当中起着非常重要的作用(李和阳等,2012),同时被普遍认为是生物活性物质和海洋药物开发的重要微生物来源(Bull et al,2005),这表明长江口及邻近海域可能蕴藏着丰富的放线菌及相关生物活性产物资源。蓝细菌是广泛分布于海洋中能进行放氧光合作用的超微型浮游植物(孙晟等,2003),是海洋初级生产力和微食物网中重要的组成成分。而在本次调查中,多个站点均发现蓝细菌且其数量不少,可以看出蓝细菌对该海域生态系统的稳定性与多样性有重要意义。
作者在实验中有发现一些分布范围和比例都较小的类群,包括厚壁菌门、绿弯菌门、酸杆菌门、浮霉菌门、疣微菌门和纤维杆菌门。厚壁菌门中的芽孢杆菌主要存在于A01和A02 站点,Vieira等(2008)指出厚壁菌丰富的地方可能表明附近海区的污染,这可能与A01和A02 两个站点处在近河口位置有关。绿弯菌是除变形菌和硝化螺旋菌外,第三类能通过硝酸盐的氧化而获得能量的一类细菌(Sorokin et al,2012),作者发现绿弯菌仅存在于近海区域受陆源输入影响而营养盐丰富的几个站点当中,因此推测绿弯菌倾向于生活在营养盐丰富的海域中。酸杆菌一般由外界环境所引入,A02 站点位于长江口受地表径流影响较大,检测到的酸杆菌很可能为陆源菌,另外Barns等(2007)曾报道过酸杆菌门类群多存在于重金属污染等酸性较强的环境中,这说明长江口区域可能存在一定程度酸化的倾向。浮霉菌门细菌属于兼性厌氧细菌,并且这类细菌可能与深海有机质的矿化有关(Sørensen et al,2005),本研究结果显示浮霉菌的相似序列来源于深海热液区。
3.3 长江口及邻近海域特殊细菌类群及分布本次实验还发现了以往同海域研究中鲜有报道的柔膜菌门和SAR 类群。目前关于柔膜菌门的报道并不多,仅侯梅锋等(2011)在青海湖底泥中有发现,主要是一类耐盐菌,另有Beazley等(2012)在墨西哥湾原油泄漏的海岸盐沼中发现大量柔膜菌的存在。本次研究在C05 站点发现少量的柔膜菌门,具体原因不详。SAR11 是最早发现的一类广泛分布于亚热带海域的浮游细菌类群,研究表明SAR11 细菌能够调控海洋中相当数量DOM 的流通,在C、N等元素的循环中起着重要的作用(Rappé et al,2002),另外海洋细菌生理代谢机制研究方面的重要发现之一是SAR11 菌群需要外源还原态硫来维持生长(Tripp et al,2008)。对SAR406类群的研究显示,它是海洋最低含氧带(Oxygen Minimum Zone,OMZ)中的优势类群(Wright et al,2012),可能与缺氧环境有关; Crump等(2007)在切萨皮克湾缺氧底层水中发现高丰度的SAR406 类群,类似于OMZ的细菌群落构成。本次研究发现A02 站点同时有SAR11和SAR406 类群的分布,可能与该站点恰好位于长江口外缺氧区,有机质和营养盐含量丰富有关。
4 结论本实验通过DAPI 染色计数和构建16S rRNA 基因文库的方法对长江口及邻近海域10 个站点表层海水中细菌的生物量及多样性进行研究。结果发现长江口及邻近海域 表层海水中细菌的丰度总体较高,但各站点间存在一定差异。海水中细菌类群主要包括变形菌门、拟杆菌门、放线菌门、蓝细菌门、厚壁菌门、绿弯菌门、酸杆菌门、浮霉菌门、纤维杆菌门、疣微菌门和柔膜菌门等11 个类群,其中α-变形菌纲所占比例最高,其次为拟杆菌门和放线菌门,其它类群的分布比例和范围都较小,同时还发现了广受关注的SAR 类群和海洋环境中少见的柔膜菌门。研究表明某些细菌类群可能成为该海域生态系统受污染的指示生物,此外未确定分类地位的细菌可能是一些未被发现的细菌种类,有待进一步调查研究。以上结果充分说明了长江口及邻近海域表层海水细菌群落结构具有丰富的多样性和复杂性。
| 王新, 李志江, 郑天凌. 2010. 海洋浮游细菌在东海赤潮高发区的分布与活性. 环境科学, 31(2): 287-295 |
| 宁修仁, 史君贤, 蔡昱明等. 2004. 长江口和杭州湾海域生物生产力锋面及其生态学效应. 海洋学报, 26(6): 96-106 |
| 刘敏, 王子峰, 朱开玲等. 2008. 应用PCR-DGGE 技术分析长江口低氧区的细菌群落组成. 高技术通讯, 18(6):650-656 |
| 刘晶晶, 杜萍, 曾江宁等. 2011. 夏季长江口浮游细菌和浮游病毒的分布特征及环境制约因素. 海洋学研究, 29(3):118-124 |
| 孙晟, 肖天, 岳海东. 2003. 秋季与春季东、黄海蓝细菌(Synechococcus spp.)生态分布特点. 海洋与湖沼, 34(2):161-168 |
| 李涛, 王鹏, 汪品先. 2008. 南海南部陆坡表层沉积物细菌和古菌多样性. 微生物学报, 48(3): 323-329 |
| 李和阳, 陈明霞, 焦念志. 2012. 长江口典型站位浮游细菌类群的组成特征研究. 海洋学报, 34(1): 183-188 |
| 张东声. 2011. 长江口及其邻近海域微生物的多样性和生态分布特征研究. 杭州: 浙江大学博士学位论文, 13-15 |
| 季倩. 2008. 长江口邻近海域水体及底栖微微型浮游生物研究. 上海: 华东师范大学硕士学位论文, 93-95 |
| 赵三军, 肖天, 岳海东. 2003. 秋季东、黄海异养细菌(Heterotrophic Bacteria)的分布特点. 海洋与湖沼, 34(3):295-305 |
| 赵海萍, 李清雪, 陶建华. 2007. 海洋细菌荧光显微计数法及其应用. 河北工程大学学报(自然科学版), 24(1): 57-60 |
| 侯梅锋, 何士龙, 李栋等. 2011. 连云港海底底泥及青海湖底泥细菌多样性研究. 环境科学, 32(9): 2681-2688 |
| 唐晓晖, 王凡. 2004. 长江口邻近海域夏, 冬季水文特征分析. 海洋科学集刊, 46: 42-66 |
| 曾永辉. 2008. 典型海洋环境中浮游细菌多样性及环境适应机制的研究. 厦门: 厦门大学博士学位论文, 34-41 |
| 潘洛安. 2005. 东海陆架及南海北部锋区微生物群落结构初探.上海: 华东师范大学硕士学位论文, 109-113 |
| Abell G C J, Bowman J P. 2005. Colonization and communitydynamics of class Flavobacteria on diatom detritus inexperimental mesocosms based on Southern Ocean seawater.FEMS Microbiology Ecology, 53(3): 379-391 |
| Barns S M, Cain E C, Sommerville L et al. 2007. Acidobacteriaphylum sequences in uranium-contaminated subsurface sedimentsgreatly expand the known diversity within the phylum. Appliedand Environmental Microbiology, 73(9): 3113-3116 |
| Beazley M J, Martinez R J, Rajan S et al. 2012. Microbialcommunity analysis of a coastal salt marsh affected by theDeepwater Horizon oil spill. PLoS One, 7(7): e41305 |
| Bowman J P, McCammon S A, Dann A L. 2005. Biogeographicand quantitative analyses of abundant uncultivatedγ-proteobacterial clades from marine sediment. MicrobialEcology, 49(3): 451-460 |
| Bowman J P, McCuaig R D. 2003. Biodiversity, communitystructural shifts, and biogeography of prokaryotes withinAntarctic continental shelf sediment. Applied andEnvironmental Microbiology, 69(5): 2463-2483 |
| Bull A T, Stach J E M, Ward A C et al. 2005. Marineactinobacteria: perspectives, challenges, future directions.Antonie van Leeuwenhoek, 87(1): 65-79 |
| Cottrell M T, Waidner L A, Yu L et al. 2005. Bacterial diversityof metagenomic and PCR libraries from the Delaware River.Environmental Microbiology, 7(12): 1883-1895 |
| Crump B C, Peranteau C, Beckingham B et al. 2007. Respiratorysuccession and community succession of bacterioplankton inseasonally anoxic estuarine waters. Applied andEnvironmental Microbiology, 73(21): 6802-6810 |
| Feng B W, Li X R, Wang J H et al. 2009. Bacterial diversity ofwater and sediment in the Changjiang estuary and coastalarea of the East China Sea. FEMS Microbiology Ecology,70(2): 236-248 |
| Inagaki F, Suzuki M, Takai K et al. 2003. Microbial communitiesassociated with geological horizons in coastal subseafloorsediments from the Sea of Okhotsk. Applied and EnvironmentalMicrobiology, 69(12): 7224-7235 |
| Jiao N Z, Zhang Y, Zeng Y H et al. 2007. Ecological anomalies inthe East China Sea: Impacts of the three gorges dam?. WaterResearch, 41(6): 1287-1293 |
| Kumar S, Tamura K, Nei M. 2004. MEGA3: Integrated softwarefor Molecular Evolutionary Genetics Analysis and sequencealignment. Briefings in Bioinformatics, 5(2): 150-163 |
| Li Y, Lee C G, Watanabe T et al. 2011. Identification of microbialcommunities that assimilate substrate from root cap cells inan aerobic soil using a DNA-SIP approach. Soil Biology andBiochemistry, 43(9): 1928-1935 |
| Lu J R, Santo Domingo J W, Lamendella R et al. 2008.Phylogenetic diversity and molecular detection of bacteria ingull feces. Applied and Environmental Microbiology, 74(13):3969-3976 |
| Mueller-Spitz S R, Goetz G W, McLellan S L. 2009. Temporal andspatial variability in nearshore bacterioplankton communities ofLake Michigan. FEMS Microbiology Ecology, 67(3): 511-522 |
| Musat F, Galushko A, Jacob J et al. 2009. Anaerobic degradationof naphthalene and 2-methylnaphthalene by strains of marinesulfate-reducing bacteria. Environmental Microbiology, 11(1):209-219 |
| Nakagawa S, Takai K, Inagaki F et al. 2005. Distribution,phylogenetic diversity and physiological characteristics ofepsilon-Proteobacteria in a deep-sea hydrothermal field.Environmental Microbiology, 7(10): 1619-1632 |
| Ning X, Lin C, Su J et al. 2011. Long-term changes of dissolvedoxygen, hypoxia, and the responses of the ecosystems in theEast China Sea from 1975 to 1995. Journal of Oceanography,67(1): 59-75 |
| Rappé M S, Connon S A, Vergin K L et al. 2002. Cultivation ofthe ubiquitous SAR11 marine bacterioplankton clade. Nature,418(6898): 630-633 |
| Ravenschlag K, Sahm K, Pernthaler J et al. 1999. High bacterialdiversity in permanently cold marine sediments. Applied andEnvironmental Microbiology, 65(9): 3982-3989 |
| Schloss P D, Handelsman J. 2005. Introducing DOTUR, acomputer program for defining operational taxonomic unitsand estimating species richness. Applied and EnvironmentalMicrobiology, 71(3): 1501-1506 |
| Sekiguchi H, Koshikawa H, Hiroki M et al. 2002. Bacterialdistribution and phylogenetic diversity in the Changjiangestuary before the construction of the Three Gorges Dam.Microbial Ecology, 43(1): 82-91 |
| Sjöstedt J, Koch-Schmidt P, Pontarp M et al. 2012. Recruitment ofmembers from the rare biosphere of marine bacterioplanktoncommunities after an environmental disturbance. Applied andEnvironmental Microbiology, 78(5): 1361-1369 |
| Sørensen K B, Canfield D E, Teske A P et al. 2005. Communitycomposition of a hypersaline endoevaporitic microbial mat.Applied and Environmental Microbiology, 71(11): 7352-7365 |
| Sorokin D Y, Lücker S, Vejmelkova D et al. 2012. Nitrificationexpanded: discovery, physiology and genomics of anitrite-oxidizing bacterium from the phylum Chloroflexi.The ISME journal, 6(12): 2245-2256 |
| Takai K, Inagaki F, Nakagawa S et al. 2003. Isolation andphylogenetic diversity of members of previously uncultivatedε-Proteobacteria in deep-sea hydrothermal fields. FEMSMicrobiology Letters, 218(1): 167-174 |
| Tripp H J, Kitner J B, Schwalbach M S et al. 2008. SAR11marine bacteria require exogenous reduced sulphur forgrowth. Nature, 452(7188): 741-744 |
| Vieira R P, Gonzalez A M, Cardoso A M et al. 2008.Relationships between bacterial diversity and environmentalvariables in a tropical marine environment, Rio de Janeiro.Environmental Microbiology, 10(1): 189-199 |
| Vörös L, Balogh K V, Koncz E et al. 2003. Phytoplankton andbacterioplankton production in a reed-covered water body.Aquatic Botany, 77(2): 99-110 |
| Wright J J, Konwar K M, Hallam S J. 2012. Microbial ecology ofexpanding oxygen minimum zones. Nature ReviewsMicrobiology, 10(6): 381-394 |
| Xu H X, Wu M, Wang X G et al. 2008. Bacterial diversity indeep-sea sediment from northeastern Pacific Ocean. ActaEcologica Sinica, 28(2): 479-485 |